
Abstract
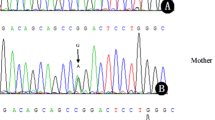
Hereditary coproporphyria (HCP) is an autosomal dominant disease characterized by a deficiency of coproporphyrinogen oxidase. To date, four mutations of the gene have been reported. We report here another mutation in two Japanese families with HCP, which was revealed by analysis of polymerase chain reaction (PCR)-amplified DNA fragments of the gene by a direct-sequencing method. A point mutation, G to A, was found in exon 4 of the gene at position 538 of the cDNA from the reported putative translation initiation codon ATG. This mutation results in a glycine to arginine substitution at amino acid 180. Two carriers in the family were successfully diagnosed by detecting the mutation using restriction analysis of the PCR products.
Similar content being viewed by others
Author information
Authors and Affiliations
Additional information
Received: 23 April 1996 / Revised: 15 July 1996
Rights and permissions
About this article
Cite this article
Daimon, M., Gojyou, E., Sugawara, M. et al. A novel missense mutation in exon 4 of the human coproporphyrinogen oxidase gene in two patients with hereditary coproporphyria. Hum Genet 99, 199–201 (1997). https://doi.org/10.1007/s004390050338
Issue Date:
DOI: https://doi.org/10.1007/s004390050338