
Abstract
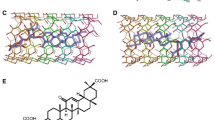
Molecular dynamics (MD) simulations for crystalline β-cyclodextrin dodecahydrate (β-CD) at two different temperatures, 293 K and 120 K, have been performed using the GROMOS program package. The calculated structural properties are compared to those obtained from neutron diffraction studies of this system at the quoted temperatures. The simulation was carried out over a period of 20 ps on four unit cells containing 8 β-CD molecules and 96 water molecules, whereby all atoms were allowed to move.
At room temperature, the experimental positions of the (non-hydrogen) glucose atoms are reproduced within 0.034 nm, a value which is smaller than the experimental (0.041 nm) or simulated (0.049 nm) overall root mean square (rms) positional fluctuation. The corresponding numbers for the low temperature study are 0.046 nm, 0.019 nm and 0.022 nm. At both temperatures the experimentally observed degree of anisotropy of the atomic motions is also found in the simulations.
The comparison of a variety of structural properties leads to the conclusion that the molecular model and force field used are able to simulate the cyclodextrin system very well. Experimentally observed differences in properties as a function of number of glucose units in the CD molecule (α-CD, 6 versus β-CD, 7) and as a function of temperature are qualitatively reproduced by the simulations.
Similar content being viewed by others

References
Atwood JL, Davies JED, MacNicol DD (1984) Inclusion compounds, vols 2, 3. Academic Press, New York
Bender ML, Komiyama M (1978) Cyclodextrin chemistry. Springer, Berlin Heidelberg New York
Betzel C, Saenger W, Hingerty BE, Brown GM (1984) Circular and flip-flop hydrogen bonding in β-cyclodextrin undecahydrate: a neutron diffraction study. J Am Chem Soc 106:7545–7556
Etten RL van, Sebastian JF, Clowes GA, Bender ML (1967) Acceleration of phenyl ester cleavage by cycloamyloses. A model for enzymatic specificity. J Am Chem Soc 89:3242–3253
Gunsteren WF van, Berendsen HJC, Hermans J, Hol WGJ, Postma JPM (1983) Computer simulation of the dynamics of hydrated protein crystals and its comparison with X-ray data. Proc Natl Acad Sci USA 80:4315–4319
Koehler JEH, Saenger W, Gunsteren WF van (1987a) A molecular dynamics simulation of crystalline α-cyclodextrin hexahydrate. Eur Biophys J 15:197–210
Koehler JEH, Saenger W, Gunsteren WF van (1987b) Molecular dynamics simulation of β-CD. The flip-flop hydrogen bonding (in preparation)
Saenger W (1980) Cyclodextrin inclusion compounds in research and industry. Angew Chem. (Int Ed Engl) 19:344–362
Saenger W, Betzel C, Hingerty BE, Brown GM (1982) Flip-flop hydrogen bonding in a partially disordered system. Nature 296:581–583
Szejtli J (1982) Cyclodextrins and their inclusion complexes. Akademiai Kiado, Budapest
Szejtli J, Fenyvesi E, Zsadon B (1978) Cyclodextrinpolymere. Stärke 30:127–131
Tutt DE, Schwartz MA (1970) Model catalysts which simulate penicillinase. V. The cycloheptaamylose — catalyzed hydrolysis of penicillins. J Am Chem Soc 93:767–771
Zabel V, Saenger W, Mason SA (1986) A neutron diffraction study of the hydrogen bonding in β-cyclodextrin undecahydrate at 120K: from dynamic flip-flops to static homodromic chains. J Am Chem Soc 108:3664–3673
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Koehler, J.E.H., Saenger, W. & van Gunsteren, W.F. Molecular dynamics simulation of crystalline β-cyclodextrin dodecahydrate at 293 K and 120 K. Eur Biophys J 15, 211–224 (1987). https://doi.org/10.1007/BF00577069
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00577069