
Abstract
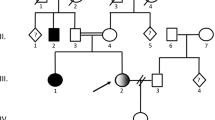
A boy and a girl born to a consanguineous Tunisian couple are suffering from a slowly progressive nervous disorder. Initially they both had normal psychomotor development with acquisition of gait and speech. First symptoms in the boy were athetoid movements during the second year of life. He later lost all motor and language skills and developed muscular rigidity and intention tremor. At the age of five years, he was completely bedridden while he appeared mentally much less affected. His younger sister followed a similar course. The major specific abnormality detected was a strikingly elevated excretion of 2-oxoglutaric acid, which was identified by gas liquid chromatography, mass spectrometry, and enzymatic analysis. 2-oxoglutarate dehydrogenase activity in homogenates of cultured skin fibroblasts was reduced to about 25% of control values in both children. Although the pathogenetic mechanisms leading to brain damage remain obscure, the finding strongly suggest an autosomal recessive neurometabolic disease with predominant involvement of the extrapyramidal system.
Similar content being viewed by others


References
Bergmeyer HU (1974) Methoden der enzymatischen Analyse. Bd. II. Verlag Chemie, Weinheim/Bergstraße
Chalmers RA, Lawson AM, Borud O (1977) Gas chromotographic and mass spectrometric studies on urinary organic acids in a patient with congenital lactic acidosis due to pyruvate decarboxylase deficiency. Clin Chim Acta 77:117–124
Chalmers RA, Ryman BE, Watts RWE (1978) Studies on a patient with in vitro evidence of type I glycogenosis and normal enzyme activities in vitro. Acta Paediatr Scand 67:201–207
Chalmers RA, Purkiss P, Watts RWE (1980) Screening for organic acidurias and amino acidopathies in newborns and children. J Inher Metab Dis 3:27–43
Christensen E, Melchior JC, Plum P (1963) Infantile Chronic Necrotizing Encephalopathy. Acta Pediatr 52:304–312
Cohen JJ (1964) Specificity of substrate utilization by the dog kidney in vivo. In: Renal metabolism and epidemiology of some renal diseases (ed J Metcalf) Maple Press, New York, pp 126–146
Finnie MDA, Cottrall K, Seakins JWT, Snedden W (1976) Massive excretion of 2-oxoglutaric acid and 3-hydroxyisovaleric acid in a patient with a deficiency of 3-methylcrotonyl-CoA carboxylase. Clin Chim Acta 73:513–519
Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC (1975) Glutaric aciduria; a “new” disorder of amino acid metabolism. Biochem Med 12:12–21
Goodman SI, Norenberg MD, Shikes RH, Breslich DJ, Moe PG (1977) Glutaric aciduria: Biochemical and morphoogical considerations. J Petiar 90:746–750
Gregersen N, Brandt NJ, Christensen E, Grøn I, Rasmussen K, Brandt S (1977) Glutaric aciduria: Clinical and laboratory findings in two brothers. J Pediatr 90:740–745
Haerer AF (1971) Citrate and α-ketoglutarate in cerebrospinal fluid and blood. Neurology 21:1059–1065
Haworth JC, Perry TL, Blass JP, Hansen S, Urquhart N (1976) Lactic acidosis in three sibs due to defects in both pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes. Pediatrics 58:564–572
Israels S, Haworth JC, Dunn HG, Applegarth DA (1976) Lactic acidosis in childhood. Adv Pediatr 22:267–303
Iversen PF, Closs K, Wad N (1971) Darkening of urine from patients treated with L-Dopa. Clin Chim Acta 32:137–139
Kippen J, Hirayama B, Klinenberg JR, Wright EM (1979) Transport of tricarboxylic acid cycle intermediates by membrane vesicles from renal brush border. Proc Natl Acad Sci USA 76:3397–3400
Kyllerman M, Steen G (1977) Intermittently progressive dyskinetic syndrome in glutaric aciduria. Neuropädiatrie 8:397–404
Langenbeck U, Hoinowski A, Mantel K, Möhring HU (1977) Quantitative gas chromatography and single-ion detection of aliphatic α-ketoacids from urine as their O-trimethylsilyl-quinoxalinol derivatives. J Chromatogr 143:39–50
Langenbeck U, Möhring HU, Hinney B, Spiteller M (1977) Quinoxalinol derivatives of aliphatic 2-oxocarboxylic acids. Infrared and mass spectra of the O-trimethylsilylated compounds. Biomed Mass Spectrom 4:197–202
Langenbeck U, Wendel U, Meuch-Hoinowski A, Kuschel D, Becker K, Przyrembel H, Bremer HJ (1978) Correlation between branched-chain amino acids and branched-chain α-ketoacids in blood in maple syrup urine disease. Clin Chim Acta 88:283–291
Lehnert W, Schuchmann L, Urbánek R, Neiderhoff H, Böhm N (1978) Excretion of 2-methyl-3-oxovaleric acid in propionic acidemia. Eur J Pediatr 128:197–205
Lowry O, Rosebrough N, Farr A, Randall RY (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Manz F, Schärer K, Alatas H, Lommel R (1974) Clearance von α-Ketoglutarat nach Stoßinjektion bei chronischer Nieren insuffizienz. Klin Wschr 52:143–144
Melançon SB, Dallaire L, Potier M (1978) Lipoamide dehydrogenase in cultured human skin fibroblasts. Clin Chim Acta 87:29–34
Saudubray JM, Dreyfus JC, Capanec C, le Lo'ch H, Pham-Hu-Trung, Mozzicanacci P (1973) Acidose lactique, hypoglycémie et hépatomégalie par deficit héréditaire en fructose-1, 6-diphosphatase hépatique. Arch Fr Pédiatr 30:609–632
Sitzmann FC (1966) Normalwerttabellen für Neugeborene, Säuglinge und ältere Kinder. 1. Teil. Pädiatr Prax 5:131
Stumpf DA, Parks IK (1978) Friedreich's ataxia: I. Normal pyruvate dehydrogenase complex activity in platelets. Ann Neurol 4:366–368
Svenningsen NW, Siensjö BK (1972) Cerebrospinal fluid lactate/pyruvate ratio in normal and asphyxiated neonates. Acta Paediatr Scand 61:117–124
Taylor SI, Mukherjee C, Jungas RL (1973) Studies on the mechanism of activation of adipose tissue pyruvate dehydrogenase by insulin. J Biol Chem 248:73–81
van Biervelieth JPGM, Bruinvis L, van der Heiden C, Ketting D, Wadman SK, Willemse JL, Monnens LAH (1977) Report of a patient with severe, chronic lactic acidemia and pyruvate carboxylase deficiency. Develop Med Child Neurol 19:392–401
van den Berg CJ, Garfinkel D (1971) A simulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J 123:211–218
Versé J (1962) Das Ketonämieproblem im Kindesalter. Klinische und experimentelle Untersuchungen. Z Kinderheilk 86:347–378
Watts RWE (1980) Neonatal screening for organic acidurias. In: Bickel H, Guthrie R, Hammersen G (eds) Neonatal screening for inborn errors of metabolism. Springer, Berlin, Heidelberg, London, pp 105–121
Wendel U, Wöhler W, Goedde HW, Langenbeck U, Passarge E, Rüdiger HW (1973) Rapid diagnosis of maple syrup urine disease(branched-chain ketoaciduria) by micro-enzyme assay in leukocytes and fibroblasts. Clin Chim Acta 45:433–440
Wiesmann UN, Rossi EE, Herschkowitz NN (1972) Correction of the defective sulfatide degradation in cultured fibroblasts from patients with metachromatic leukodystrophy. Acta Paediatr Scand 61:296–302
Author information
Authors and Affiliations
Additional information
Supported by Deutsche Forschungsgemeinschaft, SFB33
Part of the results was obtained during work for a medical thesis by G. H.
Rights and permissions
About this article
Cite this article
Kohlschütter, A., Behbehani, A., Langenbeck, U. et al. A familial progressive neurodegenerative disease with 2-oxoglutaric aciduria. Eur J Pediatr 138, 32–37 (1982). https://doi.org/10.1007/BF00442325
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00442325