
Abstract
A workshop was held on “Aspects of treatment of patients with glycogen storage disease” within the framework of the Concerted Action “Inborn errors of metabolism” of the European Communities. Consensus was reached on the main issues of treatment of patients with deficiency of glucose-6-phosphatase, glucose-6-phosphate translocase, debranching enzyme, liver phosphorylase and phosphorylase-b-kinase. The resulting recommendations are reported.
Similar content being viewed by others

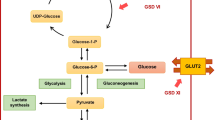
References
Bier DM, Leake RD, Haymond MW, Arnold KJ, Gruenke LD, Sperling MA, Kipnis DM (1977) Measurement of “true” glucose production rates in infancy and childhood with 6,6-dideuteroglucose. Diabetes 26:1016–1023
Chen Y-T, Cornblath M, Sidbury JB (1984) Cornstarch therapy in type I glycogen storage disease. N Engl J Med 310:171–175
Corbeel L, Boogaerts M, Van den Berghe G, Everaerts MC, Marchal G, Eeckels R (1983) Haematological findings in type Ib glycogen storage disease before and after portacaval shunt. Eur J Pediatr 140:273–275
Di Rocco M, Barrone B, Dallegri F, Frumento G, Patrone F (1984) Neutropenia and impaired neutrophil function in glycogenosis type Ib. J Inherited Metab Dis 7:151–154
Fernandes J (1974) The effect of disaccharides on the hyperlactacidaemia of glucose-6-phosphatase-deficient children. Acta paediatr Scand 63:695–698
Fernandes J, Van de Kamer JH (1968) Hexose and protein tolerance tests in children with liver glycogenosis caused by a deficiency of the debranching enzyme system. Pediatrics 41:935–944
Fernandes J, Pikaar NA (1969) Hyperlipemia in children with liver glycogen disease. Am J Clin Nutr 22:617–627
Fernandes J, Berger R (1987) Urinary excretion of lactate, 2-oxoglutarate, citrate and glycerol in patients with glycogenosis type I. Pediatr Res 21:279–282
Fernandes J, Berger R, Smit GPA (1984) Lactate as a cerebral metabolic fuel for glucose-6-phosphatase-deficient children. Pediatr Res 18:335–339
Greene HL, Slonim AE, O'Neill JA Jr, Burr IM (1976) Continuous nocturnal intragastric feeding for management of type I glycogen storage disease. N Engl J Med 294:423–425
Kraus J, Schlenker S, Schwedesky D (1974) Developmental changes of cerebral ketone body utilization in human infants. Hoppe-Seyler's Z Physiol Chem 355:164–170
Leonard JV, Dunger DB (1978) Hypoglycaemia complicating feeding regiments for glycogen storage disease. Lancet II:1203–1204
Moses SW, Gadoth N, Bashan N, Ben-David E, Slonim A, Wanderman KL (1986) Neuromuscular involvement in glycogen storage disease type III. Acta Paediatr Scand 75:289–296
Parker P, Burr I, Slonim A, Ghishan FK, Greene H (1981) Regression of hepatic adenomas in type la glycogen storage disease with dietary therapy. Gastroenterology 81:534–536
Schaub J, Heyne K (1983) Glycogen storage disease type Ib. Eur J Pediatr 140:283–288
Slonim AE, Coleman RA, Moses SW (1984) Myopathy and growth failure in debrancher enzyme deficiency: improvement with high-protein nocturnal enteral therapy. J Pediatr 105:906–911
Stanley CA, Mills JL, Baker L (1981) Intragastric feeding in type I glycogen storage disease: factors affecting the control of lactic acidemia. Pediatr Res 15:1504–1508
Zangeneh F, Limbeck GA, Brown BI, Emch JR, Arcasoy MM, Goldenberg VE, Kelley VC (1969) Hepatorenal glycogenosis (type I glycogenosis) and carcinoma of the liver. J Pediatr 74:73–83
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Fernandes, J., Leonard, J.V., Moses, S.W. et al. Glycogen storage disease: recommendations for treatment. Eur J Pediatr 147, 226–228 (1988). https://doi.org/10.1007/BF00442683
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00442683