
Abstract
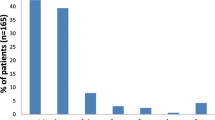
3-Methylglutaconic aciduria has been found in two distinct syndromes. In one there is deficient activity of 3-methylglutaconyl coenzyme A hydratase, and the only clinical manifestation observed has been retardation of speech development. In the other, which includes a majority of the patients studied, we document that the activity of this enzyme in fibroblast extracts is normal. The phenotype of this disorder is one of profound neurological impairment with retarded psychomotor development, hypotonicity and/or spasticity, convulsions or EEG abnormalities, and sensorineural changes in the eye and ear.
Similar content being viewed by others

Abbreviations
- 3-MG-CoA:
-
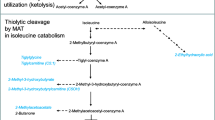
3-methylglutaconyl coenzyme A
- PAS:
-
periodic acid-Schiff
- ATP:
-
adenosine-5-triphosphate
- HMG:
-
3-hydroxy-3-methylglutaric acid
- 3-HB:
-
3-hydroxybutyric acid
- EDTA:
-
ethylene-diaminetetra-acetic acid
References
Divry P, Vianey-Liaud C, Mory O, Ravussin JJ (1987) 3-Methylglutaconic aciduria: familial neonatal form with fatal onset. J Inherited Metab Dis [Suppl 2] 10:286–289
Duran M, Beemer FA, Tibosch AS, Bruinvis L, Ketting D, Wadman SK (1982) Inherited 3-methylglutaconic aciduria in two brother — another defect of leucine metabolism. J Pediatr 101: 551–554
Edmond J, Popjak G (1974) Transfer of carbon atoms from mevalonate to n-fatty acids. J Biol Chem 249:66–71
Gibson KM, Sweetman L, Nyhan WL, Narisawa K, Roth K, Lehnert W, Robinson B, Duran M, Wadman SK (1985) 3-Methylglutaconyl-CoA hydratase deficiency: two different clinical and enzymatic phenotypes in 3-methylglutaconic aciduria. Pediatr Res 19:248a
Greene CL, Cann HM, Robinson BH, Gibson KM, Sweetman L, Holm J, Nyhan WL (1984) 3-Hydroxy-3-methylglutaric aciduria. J Neurogenet 1:165–173
Greter J, Hagberg B, Steen G, Söderhjelm U (1978) 3-Methylglutaconic aciduria: report on a sibship with infantile progressive encephalopathy. Eur J Pediatr 129:231–238
Haan EA, Scholem RD, Pitt JJ, Wraith JE, Brown GK (1987) Episodes of severe metabolic acidosis in a patient with 3-methylglutaconic aciduria. Eur J Pediatr 146:484–488
Hagberg B, Hjalmarson O, Lindstedt S, Ransnäs L, Steen G (1983) 3-Methylglutaconic aciduria in two infants. Clin Chim Acta 134:59–67
Hammond J, Wilcken B (1984) 3-Hydroxy-3-methylglutaric, 3-methylglutaconic and 3-methylglutaric acids can be non-specific indicators of metabolic disease. J Inherited Metab Dis [Suppl 2] 7:117–118
Landaas S (1974) Increased urinary excretion of 3-hydroxyisovaleric acid in patients with ketoacidosis. Clin Chim Acta 54:39–46
Lehnert W, Scharf J, Wendel U (1985) 3-Methylglutaconic and 3-methylglutaric aciduria in a patient with suspected 3-methylglutaconyl-CoA hydratase deficiency. Eur J Pediatr 143:301–303
Narisawa K, Gibson KM, Sweetman L, Nyhan WL, Duran M, Wadman SK (1986) Deficiency of 3-methylglutaconyl-coenzyme A hydratase in two siblings with 3-methylglutaconic aciduria. J Clin Invest 77:1148–1152
Robinson BH, Sherwood WG, Lampty M, Lowden JA (1976) β-Methylglutaconic aciduria: a new disorder of leucine metabolism. Pediatr Res 10:371
Steen G, Ransnäs L (1983) Organic acids of urine in multiple sclerosis. Acta Neurol Scand 68:231–240
Sweetman L (1984) Qualitative and quantitative analysis of organic acids in physiologic fluids for diagnosis of the organic acidurias. In: Nyhan WL (ed) Abnormalities in amino acid metabolism in clinical medicine. Appleton-Century-Crofts, Norwalk, Conn, pp 419–453
Truscott RJW, Halpern B, Wysocki SJ, Haehnel R, Wilcken B (1979) Studies on a child suspected of having a deficiency in 3-hydroxy-3-methylglutaryl-CoA lyase. Clin Chim Acta 95:11–16
Wiley MH, Howton MM, Siperstein MD (1979) Sex differences in the sterol and nonsterol metabolism of mevalonate. J Biol Chem 254:837–842
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gibson, K.M., Nyhan, W.L., Sweetman, L. et al. 3-Methylglutaconic aciduria: a phenotype in which activity of 3-methylglutaconyl-coenzyme A hydratase is normal. Eur J Pediatr 148, 76–82 (1988). https://doi.org/10.1007/BF00441821
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00441821