
Abstract
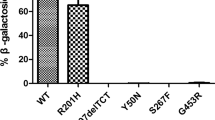
We have studied, by the polymerase chain reaction, the β-galactosidase cDNA from several Italian patients with infantile GM1-gangliosidosis. One homozygote for a previously undiscovered G > A mutation at position 1479, causing an arginine to histidine change, was detected. The same mutation, in heterozygosis, was identified in 6 unrelated patients, but not in 100 normal chromosomes.
Similar content being viewed by others

References
Arcari P, Martinelli R, Salvatore F (1984) The complete sequence of a full length cDNA for human liver glyceraldehyde-3-phosphate dehydrogenase: evidence for multiple mRNA species. Nucleic Acids Res 12:9179–9189
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Conzelmann E, Sandhoff K (1987) Glicolipid and glicoprotein degradation. Adv Enzymol 60:89–217
Crotta S, Nicolis S, Ronchi A, Ottolenghi S, Ruzzi L, Shimada Y, Migliaccio AR, Migliaccio G (1990) Progressive inactivation of the expression of an erythroid transcriptional factor in GM and G-CSF-dependent myeloid cell lines. Nucleic Acids Res 18:6863–6869
Groebe H, Krins M, Schmidberger H, Figura K von, Harzer K, Kresse H, Paschke E, Sewell A, Ullrich K (1980) Morquio syndrome (mucopolysaccharidosis IV-B) associated with β-galactosidase deficiency. Report of two cases. Am J Hum Genet 32:258–272
Morreau H, Galjart NJ, Gillemans N, Willemsen R, Horst GTJ van der, d'Azzo A (1989) Alternative splicing of β-galactosidase mRNA generates the classic lysosomal enzyme and a βgalactosidase-related protein. J Biol Chem 264:20655–20663
Nishimoto J, Nanba E, Inui K, Okada S, Suzuki K (1991) GM1gangliosidosis (genetic β-galactosidase deficiency): identification of four mutations in different clinical phenotypes among Japanese patients. Am J Hum Genet 49:566–574
O'Brien JS (1989a) The gangliosidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 945–972
O'Brien JS (1989b) β-galactosidase deficiency (GM1-gangliosidosis, galactosialidosis, and Morquio syndrome type B); ganglioside sialidase deficiency (mucolipidosis IV). In: Scriver CR, Beaudet AL. Sly WS, Val'le D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1797–1806
Okada S, O'Brien JS (1968) Generalized gangliosidosis: β-galactosidase deficiency. Science 160:1002–1004
Oshima A, Tsuji A, Nagao Y, Sakuraba H, Suzuki Y (1988) Cloning, sequencing, and expression of cDNA for human β-galactosidase. Biochem Biophys Res Commun 157:238–244
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci USA 74:294–5299
Yoshida K, Oshima K, Shimmoto M, Fukuhara Y, Sakuraba H, Yanagisawa N, Suzuki Y (1991) Human β-galactosidase gene mutations in GM1-gangliosidosis: a common mutation among Japanese adult chronic cases. Am J Hum Genet 49:435–442
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Mosna, G., Fattore, S., Tubiello, G. et al. A homozygous missense arginine to histidine substitution at position 482 of the β-galactosidase in an Italian infantile GM1-gangliosidosis patient. Hum Genet 90, 247–250 (1992). https://doi.org/10.1007/BF00220071
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00220071