
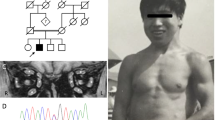
Summary
A 40-year-old man suffered for 5 years from a progressive proximal myopathy mimicking an atypical limb-girdle dystrophy. A “myopathic” pattern with myotonic and pseudomyotonic discharges was determined by electromyography. Enzyme histochemical and ultrastructural investigations of muscle and liver biopsies pointed to a glycogenosis. Biochemical investigations of muscle and liver samples confirmed this diagnosis, disclosing an acid maltase deficiency. Glycogen filled lysosomes were also revealed electron optically in skin fibroblasts but not in white blood cells.
The literature concerning the late onset forms of acid maltase deficiency (type II glycogenosis) has been reviewed, and the clinical course has been compared with that of the infantile form (Pompe's disease). In early infancy the disease has a short and fatal course, with involvement of many organs, primarily skeletal muscles, liver and heart. In the late infantile and juvenile forms the course of the disease is slower, the organ involvement beeing not as severe; muscular symptoms begin to prevail. In adults, type II glycogenosis mimics muscular dystrophy with its prolonged course and the almost exclusive clinical involvement of proximal muscles. Biochemical and ultrastructural investigations have nevertheless demonstrated that other organs and tissues are also involved. The reasons for the variability of organ involvements in different ages are as yet unknown.
Zusammenfassung
Bericht über einen 40jährigen Patienten, der seit 5 Jahren an einer progredienten Myopathie litt und neurologisch das Bild einer atypischen Beckengürteldystrophie bot. Im EMG wurden myotone und pseudomyotone Entladungen beobachtet. Die enzymhistochemischen und elektronenoptischen Untersuchungen der Muskelbiopsien und eines Leberpunktates wiesen auf eine Glykogenose hin; die biochemische Untersuchung bestätigte den Mangel an saurer Maltase.
Weitere Literaturfälle von Saure-Maltase-Mangel-Syndrom im juvenilen und Erwachsenenalter wurden überprüft. Es stellt sich heraus, daß mit zunehmendem Alter des Patienten ein fast selektiver Befall der proximalen Muskeln auftritt, was oft Anlaß zur Verwechslung mit anderen sog. degenerativen neuromuskulären Erkrankungen gibt. Bioptische und biochemische Untersuchungen sind daher in solchen Fällen besonders indiziert. Der langsamere Verlauf dieser generalisierten Speicherkrankheit im Erwachsenenalter — gegenüber der rasch verlaufenden infantilen Form (Pompesche Erkrankung) — bleibt ungeklärt.
Similar content being viewed by others


Literatur
Angelini, C., Engel, A. G.: Comparative study of acid maltase deficiency. Biochemical differences between infantile, childhood, and adult types. Arch. Neurol. 26, 344–349 (1972)
Badoual, J., Lestradet, H., Vilde, J. L., Ploussard, J. P.: Une forme atypique de glycogénose par déficit en maltase acide. Sem. Hôp. Paris 43, 1427–1434 (1967)
Canal, L., Frattola, L., Pellegrini, G.: Skeletal muscle glycogenosis type II: Biochemical and electron microscopic investigations of one case. Z. Neurol. 201, 98–108 (1972)
Carrier, H., Lebel, M., Mathieu, M., Pialat, J., Devic, M.: Late familial pseudo-myopathic muscular glycogenosis with alpha 1,4 glucosidase deficiency. Morphological, histoenzymological and biochemical approach. Path. europ. 10, 51–59 (1975)
Courtecuisse, V., Royer, P., Habib, R., Monnier, C., Demos, J.: Glycogénose musculaire par déficit en alpha 1–4 glucosidase simultant una dystrophie musculaire progressive. Etude clinique et enzymatique, microscopie optique et électronique. Arch. franç. Pédiat. 22, 1153–1164 (1965)
De Barsy, Th., Van Hoof, F., Ermel, A. E., Brucher, J. M., Billiet, L.: Adult form of acid maltase deficiency: Biochemical and neuropathological study of two siblings. Proc. VIIth Intern. Congr. Neuropathology. Budapest: Kultura 1975
Dreyfus, J.-Cl.: Les glycogénoses musculaires. Etude biochimique. Ann. Anat. Path. 18, 169–194 (1973)
Engel, A. G.: Acide maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain 93, 599–616 (1970)
Engel, A. G.: Vacuolar myopathies. Multiple etiologies and sequential structural studies. In: The striated muscle (C. M. Pearson, F. K. Mostofi, eds.). Baltimore: The Williams & Wilkins Co. 1973
Engel, A. G., Dale, A.-D.: Autophagic glycogenosis of late onset with mitochondrial abnormalities: light and electron microscopic observations. Mayo Clin. Proc. 43, 233–279 (1968)
Engel, A. G., Gomez, M. R., Seybold, M. E., Lambert, E. H.: The spectrum and diagnosis of acid maltase deficiency. Neurology (Minneap.) 23, 95–106 (1973)
Erbslöh, F.: Progressive Myopathien bei Glykogenosen. In: G. Bodechtel, Differentialdiagnose neurologischer Krankheitsbilder, 3. Aufl., S. 884. Stuttgart: Thieme 1974
Gulotta, F., Payk, Th. R., Solbach, A.: Sudanophile (mitochondriale) Myopathie. Z. Neurol. 206, 309–326 (1974)
Hers, H. G.: Alpha-Glucosidase deficiency in generalized glycogen storage disease (Pompe's disease). Biochem. J. 86, 11–16 (1963)
Hers, H. G.: The concept of inborn lysosomal disease. In: Lysosomes and storage diseases (H. G. Hers, F. Van Hoof, eds.), pp. 148–172. New York-London: Academic Press Inc. 1973
Hers, H. G., de Barsy, Th.: Type II glycogenosis (acid maltase deficiency). In: Lysosomes and storage disease (H. G. Hers, F. Van Hoof, eds.), pp. 197–217. New York-London: Academic Press Inc. 1973
Hudgson, P., Gardner-Medwin, D., Worsfold, M., Pennington, R. J. T., Walton, J. N.: Adult myopathy from glycogen storage disease due to acid maltase deficiency. Brain 91, 435–462 (1968)
Isch, F., Juif, J.-G., Sacrez, R., Thiebaut, F.: Glycogénose musculaire a forme myopathique par déficit en maltase acide. Pédiatrie 21, 71–86 (1966)
Kölmel, H. W., Assmus, H., Seiler, D.: Myopathie bei Saure-Maltase-Mangel. Die Pompesche Erkrankung im Jugend- und Erwachsenenalter. Arch. Psychiat. Nervenkr. 218, 93–106 (1974)
Lenard, H. G., Schaub, J., Keutel, J., Osang, M.: Electromyography in type II glycogenosis. Neuropädiatrie 5, 410–424 (1974)
Martin, J. J., de Barsy, Th., Van Hoof, F., Palladini, G.: Pompe's disease: An inborn lysosomal disorder with storage of glycogen. A study of brain and striated muscle. Acta neuropath. (Berl.) 23, 229–244 (1973)
Nevsîmal, O., Kocura, P.: Mîrný průbeh generalizované glykogenózy (Morbus Pompe). Čs. Neurol. Neurochir. 36, 174–178 (1973)
Schotland, D. L.: Ultrastructure of muscle in glycogen storage disease. In: The striated muscle (C. M. Pearson, F. K. Mostofi, eds.), pp. 410–426. Baltimore: The Williams & Wilkins Co. 1973
Sengel, A., Stoebner, P., Isch, F.: Une myopathie vacuolaire: La glycogénose autophagique à début tardif. Etude ultrastructurale. Ann. Anat. Path. 16, 47–54 (1971)
Smith, H. L., Amick, L. D., Sidbury, J. B. Jr.: Type II glycogenosis: Report of a case with four year survival and absence of acid maltase associated with an abnormal glycogen. Amer. J. Dis. Child. 111, 475–481 (1966)
Smith, J., Zellweger, H., Afifi, A. K.: Muscular form of glycogenosis type II (Pompe): Report of a case with unusual features. Neurology (Minneap.) 17, 537–549 (1967)
Swaiman, K. F., Kennedy, W. R., Sauls, H. S.: Late infantile acid maltase deficiency. Arch. Neurol. 18, 642–648 (1968)
Zellweger, H., Illingworth Brown, B., McCormick, W. F., Tu, J.-B.: A mild form of muscular glycogenosis in two brothers with alpha-1,4-glucosidase deficiency. Ann. paediat. (Basel) 205, 413–437 (1965)
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gullotta, F., Stefan, H. & Mattern, H. Pseudodystrophische Muskelglykogenose im Erwachsenenalter (Saure-Maltase-Mangel-Syndrom). J. Neurol. 213, 199–216 (1976). https://doi.org/10.1007/BF00312870
Received:
Issue Date:
DOI: https://doi.org/10.1007/BF00312870